단백질 구조 예측의 난제, AlphaFold 2가 어떻게 해결했는지 궁금하시죠? CASP14에서 압도적인 성능을 보인 AlphaFold 2의 혁신적인 아키텍처와 원자 수준 정확도의 원리를 학습 노트에서 함께 살펴봅니다. 생명 과학의 미래를 바꾼 이 기술의 핵심을 지금 바로 확인하세요!
AlphaFold 2: CASP14를 정복한 딥러닝 혁명! 원자 수준 단백질 구조 예측의 놀라운 9가지 비밀.
안녕하세요! 제가 이번에 존 점퍼(John Jumper)와 딥마인드 팀이 발표한 그 유명한 논문, 'AlphaFold를 이용한 매우 정확한 단백질 구조 예측 (Highly accurate protein structure prediction with AlphaFold: AlphaFold)'을 정말 깊이 있게 읽어보았는데요. 솔직히 말해서, 이 논문은 생명 과학 분야에서 역사적인 이정표를 세운 것 같아요. 저처럼 생물정보학에 관심 있는 분들이라면 이 혁명이 대체 어떤 원리로 가능한 건지, 왜 그렇게 대단한 일인지 궁금하실 거예요.
단백질의 3차원 구조를 예측하는 문제, 소위 '단백질 접힘 문제(Protein Folding Problem)'는 무려 50년 이상 풀리지 않던 난제였잖아요? 실험적으로 단백질 구조 하나를 결정하는 데는 짧게는 몇 달, 길게는 몇 년의 고된 노력이 필요했는데, 그 속도에 비해 알려진 단백질 서열은 수십억 개에 달하니 그 간극이 정말 엄청났죠. 그런데 AlphaFold가 등장하면서 이 간극을 거의 원자 수준의 정확도로 메우기 시작했다는 건, 연구자로서 정말 꿈만 같은 일인 것 같아요. 이 글에서는 제가 논문을 읽으면서 느꼈던 충격과 감동을 그대로 담아, 이 기술의 핵심 논리와 과학적 배경을 저의 학습 노트에서 자세하게 풀어보려고 합니다. 자, 그럼 이 놀라운 여정을 함께 떠나볼까요?
1. AlphaFold, 단백질 구조 예측의 새로운 시대 (Abstract)
논문의 초록부터 제 눈을 사로잡은 문구는 바로 이것이었어요. "우리는 유사한 구조가 전혀 알려지지 않은 경우에도 정기적으로 단백질 구조를 원자 수준의 정확도로 예측할 수 있는 최초의 계산 방법을 제공한다." 이 말은 그동안 우리가 알던 단백질 구조 예측의 한계를 완전히 뛰어넘었다는 것을 의미하거든요. 이전에 아무리 발전해도 원자 수준의 정확도에는 미치지 못했고, 특히 상동성이 없는 (Homologous structure is not available) 단백질의 구조를 예측하는 것은 거의 불가능에 가까웠잖아요. 그런데 이 논문은 새롭게 재설계된 AlphaFold가 이 문제를 해결했다고 선언하고 있어요.
저도 CASP14 결과를 보고 정말 깜짝 놀랐는데요. CASP14(Critical Assessment of protein Structure Prediction)는 단백질 구조 예측 분야의 올림픽 같은 대회인데, AlphaFold는 거기서 대부분의 경우 실험 구조와 경쟁할 만한 정확도를 보여주었대요. 다른 기존 방법들을 압도적으로 능가했다는 사실이 논문 초록에서부터 명확하게 드러나더라고요. 이전 버전의 AlphaFold도 훌륭했지만, 이 논문에서 다루는 버전은 완전히 재설계된 신경망 기반 모델이랍니다. 특히 CASP14 평가에서는 실험 구조와 구별하기 어려울 정도의 정확성을 달성한 경우가 많았다고 하니, 이게 얼마나 엄청난 발전인지 실감하게 되죠.
이 기술의 핵심은 단순한 딥러닝 알고리즘이 아니라는 점이 중요해요. 논문 초록을 자세히 읽어보면, AlphaFold의 최신 버전은 단백질 구조에 대한 물리적 및 생물학적 지식을 통합한 새로운 기계 학습 접근 방식을 기반으로 하고 있다는 것을 알 수 있어요. 특히 다중 서열 정렬(Multi-Sequence Alignments, MSA) 정보를 딥러닝 알고리즘 설계에 적극적으로 활용했다는 점이 혁신의 중심에 있는 것 같더라고요. MSA는 단백질의 진화적 역사를 담고 있는 중요한 정보인데, 이것을 신경망에 통합하는 방식 자체가 기존의 예측 방법들과는 궤를 달리하는 접근이었죠. 이 혁신 덕분에 수십 년간 생물학계의 숙원이었던 '단백질 접힘 문제'의 계산적 해결 가능성이 눈앞에 펼쳐지게 된 것이라고 저자는 이야기하고 있답니다. 단순히 정확도를 높인 정도가 아니라, 이 분야의 패러다임을 바꾼 사건이라고 평가할 수밖에 없는, 그런 내용으로 초록이 꽉 채워져 있었어요. 논문 전체를 읽기 전부터 기대감이 폭발하더라고요.
AlphaFold의 등장은 단순히 구조 예측의 정확도를 높인 것을 넘어, 구조 생물정보학(Structural Bioinformatics)을 대규모로 가능하게 할 기반을 마련했다는 데 큰 의의가 있어요. 이전까지 몇 년씩 걸리던 과정을 순식간에 해낼 수 있게 되었으니, 이제 수많은 단백질 서열에 대한 구조적 커버리지를 넓히는 것이 가능해진 거죠. 이 논문이 발표됨으로써 우리는 단백질의 기능에 대한 기계론적 이해(mechanistic understanding of their function)를 촉진하고, 새로운 약물 발견이나 효소 설계와 같은 응용 분야에 엄청난 가속도를 붙일 수 있게 되었다고 저자들은 자신 있게 주장하고 있답니다. 이 모든 내용이 초록 단 세 단락에 압축되어 있다는 것이 정말 놀라웠어요.
저자가 이렇게 자신감을 가질 수 있었던 배경에는 CASP14에서의 독보적인 성능이 있었을 거예요. 이전에 다른 방법들이 '원자 수준 정확도'에는 훨씬 미치지 못했던 것과 달리, AlphaFold는 그 벽을 허물었다는 거죠. 특히 '상동 구조가 알려져 있지 않은 경우(no similar structure is known)'에도 높은 정확도를 보였다는 부분은, 우리가 오랫동안 의존해왔던 템플릿 기반 모델링(Template-based modeling)의 한계를 뛰어넘었다는 것을 의미합니다. 이 부분이 바로 AlphaFold의 기술적 성숙도와 혁신성을 가장 잘 보여주는 핵심 포인트라고 저는 생각해요. 이 기술 덕분에 수많은 생명 현상의 비밀이 단백질 구조를 통해 더 빨리 밝혀질 것이라는 기대감이 저절로 생기지 않나요? 정말 흥미로운 시작이었어요.
2. 풀리지 않던 난제, 단백질 접힘 문제와 기존 방법의 한계 (Introduction)
서론 부분은 이 논문이 왜 필요한지, 그리고 단백질 구조 예측 문제가 얼마나 중요하고 어려웠는지를 명확하게 설명해 주더라고요. 단백질은 생명의 기본 요소이고, 그 기능을 이해하려면 당연히 3차원 구조를 알아야 합니다. 우리가 지금까지 엄청난 실험적 노력으로 약 10만 개 정도의 고유 단백질 구조를 밝혀냈지만, 이는 알려진 수십억 개의 단백질 서열에 비하면 정말 작은 조각에 불과하대요. 서열은 너무나 빨리 쌓이는데, 구조를 밝히는 속도가 너무 느렸던 거죠.
저자들은 구조적 커버리지(Structural coverage)가 '하나의 단백질 구조를 결정하는 데 필요한 수개월에서 수년에 걸친 고통스러운 노력' 때문에 병목 현상을 겪고 있다고 지적합니다. 저도 그 어려움을 어느 정도 알기 때문에, 이 병목을 해소할 정확한 계산 접근 방식이 절실했다는 데 크게 공감했어요. 여기서 다시 한번 '단백질 접힘 문제(Protein folding problem)'가 등장하는데, 이는 아미노산 서열만으로 단백질이 어떤 3차원 구조를 가질지 예측하는 것이었고, 50년 넘게 미해결된 중요한 연구 과제였죠.
서론에서는 기존의 계산 방법을 크게 두 가지 상보적인 경로로 분류하고 설명해 주는데, 이 부분이 정말 깔끔하게 정리되어 있어서 좋았습니다. 하나는 물리적 상호작용(Physical interactions)에 초점을 맞춘 경로, 다른 하나는 진화적 역사(Evolutionary history)에 초점을 맞춘 경로입니다. 이 두 경로가 각각 어떤 한계를 가졌는지 명확히 짚어주더라고요.
첫 번째, 물리적 상호작용 프로그램은 분자 구동력에 대한 이해를 바탕으로 단백질 물리학의 열역학적 또는 운동학적 시뮬레이션에 중점을 둡니다. 이론적으로는 가장 매력적인 접근법이지만, 실제로 해보려니 너무 어려웠던 거죠. 분자 시뮬레이션의 계산적 난해성(computational intractability), 단백질 안정성의 맥락 의존성(context dependence), 그리고 충분히 정확한 단백질 물리학 모델을 만들기가 어렵다는 문제 때문에 중간 크기 단백질조차도 예측하기가 매우 어려웠다고 저자들은 설명합니다. 아무리 고성능 컴퓨터가 있어도, 자연의 복잡한 물리 법칙을 완벽하게 시뮬레이션하는 건 정말이지 벽처럼 느껴지는 일이었을 거예요.
두 번째, 진화적 프로그램은 최근 몇 년 동안 대안을 제시해 왔습니다. 이 접근법은 단백질의 진화적 역사에 대한 생물정보학적 분석, 풀린 구조와의 상동성(homology), 그리고 쌍별 진화적 상관관계(pairwise evolutionary correlations)에서 구조적 제약 조건을 도출하는 방식이었죠. 이 접근법은 PDB(Protein Data Bank)에 실험 단백질 구조가 꾸준히 증가하고, 게놈 염기서열 분석이 폭발적으로 늘어나면서 큰 도움을 받았어요. 특히 쌍별 상관관계(Co-evolution)를 딥러닝과 결합한 방법들이 CASP13 이전부터 괄목할 만한 발전을 이루긴 했어요. 하지만 저자들은 이러한 최근의 발전에도 불구하고, 기존 방법들은 '원자 정확도(atomic accuracy)'에는 여전히 크게 미치지 못했다는 점을 명확히 합니다. 특히 상동 구조를 찾기 어려운 경우 (이른바 'Free Modeling' 혹은 'De Novo' 타겟)에는 그 한계가 더욱 뚜렷했고요.
이러한 상황에서, 논문의 저자들은 AlphaFold가 기존 방법들이 가지는 구조적 정보 통합의 한계를 뛰어넘어, 진화적 정보를 딥러닝 알고리즘 내부에 더 깊이, 그리고 더 효과적으로 통합하는 새로운 길을 제시했다는 것을 서론에서부터 강조하고 있습니다. 이 접근법 덕분에 오랫동안 해결 불가능해 보였던 '상동성 없는 단백질 구조 예측'이라는 난제를 해결할 수 있었다는 거죠. 서론을 읽으면서, 이 논문이 단순한 기술 개선이 아니라, 기초 생물학 난제에 대한 근본적인 해결책을 제시하고 있다는 확신을 얻을 수 있었답니다. 정말 짜릿한 시작이라고밖에는 표현할 수 없을 것 같아요!
3. CASP14를 압도하다: AlphaFold 2의 경이로운 예측 결과 (Results)
가장 흥미로운 부분 중 하나는 바로 결과(Results) 섹션이었어요. CASP14에서의 성능을 자세히 분석했는데, AlphaFold의 구조는 경쟁 방법들보다 훨씬 더 정확했다고 저자들은 자신 있게 보고하고 있답니다. 특히 이 논문에서는 CASP14의 메인 평가 지표인 GDT-TS(Global Distance Test - Total Score)를 중심으로 설명하고 있는데, GDT-TS 점수는 100점에 가까울수록 실험 구조와 거의 일치한다는 것을 의미합니다. CASP14의 전체 타겟을 대상으로 한 평균 GDT-TS 점수에서 AlphaFold는 다른 팀들을 압도적으로 따돌렸다고 해요.
저자들은 AlphaFold가 특히 예측이 어려웠던 '자유 모델링(Free Modeling, FM)' 타겟에서도 훌륭한 성능을 보여주었다는 점을 강조합니다. FM 타겟은 기존에 알려진 구조적 상동성이 거의 없는 단백질을 예측해야 하므로, 사실상 '맨땅에 헤딩'하는 수준의 난제였거든요. 그런데 AlphaFold는 이 FM 타겟에서도 GDT-TS 점수 80점을 넘기는 등, 실험적으로 결정된 구조와 비교해도 손색이 없는 정확도를 달성했다는 것이 정말 놀랍습니다. 저자들의 관점에서는, 이 결과가 단백질 구조 예측 문제가 사실상 해결되었다고 볼 수 있는 중요한 근거 중 하나가 됩니다.
단순히 GDT-TS 점수뿐만 아니라, 예측 구조의 원자 수준 정확성(atomic-level accuracy)을 확인하기 위한 분석도 진행되었어요. CASP14에서는 글로벌 코디네이트 에러(Global coordinate error)를 측정했는데, AlphaFold 예측 구조의 평균 에러는 실험적으로 결정된 구조 간의 차이와 비슷한 수준이었다고 합니다. 즉, AlphaFold가 예측한 구조와 실제 실험 구조의 차이가, 하나의 단백질을 두 번 다른 실험 방법으로 측정했을 때 나오는 차이만큼 작았다는 것을 의미해요. 이 말은 곧, AlphaFold의 예측 구조를 실제로 실험적으로 검증된 구조처럼 신뢰할 수 있다는 뜻이 되겠죠. 저도 이 결과를 보면서 정말 감격했습니다. 이게 바로 '원자 수준 정확도'를 달성했다는 주장의 핵심적인 뒷받침이 되는 데이터인 거죠.
논문에는 AlphaFold가 특히 정확도가 낮았을 것으로 예상되는 어려운 영역, 예를 들어 긴 루프(long loops)나 불규칙한 구조(irregular motifs)에서도 경쟁자들보다 훨씬 뛰어난 성능을 보였다는 내용이 포함되어 있어요. 루프 같은 유연한 영역은 예측하기가 정말 까다로운데, AlphaFold는 이런 부분까지도 정확하게 잡아냈다는 것은 모델이 단백질의 국소적인 상호작용과 전체적인 구조적 제약을 동시에 이해하고 있음을 시사하는 결과랍니다. 또한, 논문은 최근 PDB에 등록된 구조를 대상으로 한 자체적인 검증 데이터셋에서도 AlphaFold가 훌륭한 성능을 유지했음을 보여주는데, 이는 모델의 일반화 능력(Generalization ability)이 매우 뛰어나다는 것을 입증하는 것이라고 볼 수 있습니다.
결과적으로, AlphaFold는 CASP14를 통해 단백질 구조 예측 분야에서 기존의 모든 예측 방법론을 구시대의 유물로 만들어버리는 압도적인 결과를 보여주었어요. 이 성공은 MSA 정보와 단백질 구조에 대한 깊은 생물학적 통찰력을 딥러닝 아키텍처에 성공적으로 통합한 결과이며, 앞으로 수많은 생물학 연구를 가속화할 것이라는 저자들의 주장이 전혀 과장이 아니라는 것을 수많은 데이터와 수치들이 증명하고 있답니다. 이 결과를 보면서 저는 이 논문이 생물학계에 미칠 파급 효과에 대해 다시 한번 깊이 생각하게 되었어요. 정말이지 놀라운 성과입니다!
4. 데이터로 확인하는 AlphaFold의 정확성: 그림과 CASP14 지표 분석
논문의 결과 섹션은 글로만 읽어서는 그 엄청난 차이를 실감하기 어려워요. 당연히 논문에는 예측 구조와 실험 구조를 시각적으로 비교한 그림들이 포함되어 있는데요. 그 그림들을 보면 AlphaFold가 예측한 구조가 실험적으로 밝혀진 구조 위에 마치 그림자처럼 정확히 겹쳐져 있는 것을 볼 수 있답니다. 기존의 방법들은 대체로 전체적인 형태만 비슷하고 세부적인 측면에서는 많은 오류를 보였지만, AlphaFold는 개별 아미노산 잔기(residue)의 위치까지도 거의 완벽하게 재현해 냈어요.

CASP14에서 사용된 주요 평가 지표를 좀 더 자세히 뜯어보면, AlphaFold의 독보적인 성능이 수치로 명확하게 드러납니다. 앞서 언급한 GDT-TS는 구조적 유사성을 측정하는 가장 일반적인 지표인데, AlphaFold는 경쟁 2등 팀보다도 훨씬 높은 평균 점수를 기록했어요. 이 차이는 통계적으로 매우 유의미하며, 단순히 약간의 개선이 아니라 근본적인 성능 도약을 의미하는 거였죠. 논문 그림에는 이 점수가 다른 예측 방법들과 비교되어 막대 그래프나 분포도로 제시되는데, 그 차트의 압도적인 높이를 보면 정말 입이 다물어지지 않습니다.
특히 저자들이 강조하는 부분은, pLDDT(predicted Local Distance Difference Test)라는 자체적인 신뢰도 점수의 유용성이에요. 이 지표는 예측된 구조의 각 잔기(residue)별로 예측 정확도를 평가하는 점수인데, AlphaFold는 이 pLDDT 점수를 통해 자신이 예측한 구조의 어느 부분이 신뢰할 만한지, 아니면 불확실한지를 사용자에게 알려줄 수 있었답니다. 예를 들어, pLDDT 점수가 높은 영역은 실제 실험 구조와 거의 일치하는 반면, 점수가 낮은 영역은 유연하거나 무질서한(disordered) 영역일 가능성이 높다는 것을 알 수 있죠. 이처럼 예측의 정확성뿐만 아니라 예측된 구조의 신뢰도까지 함께 제공한다는 것은, 이 기술을 실제 생물학 연구에 활용하는 사람들에게는 정말 엄청난 이점이 아닐 수 없어요.
논문 그림에는 AlphaFold 아키텍처의 도식(Model Architecture Schematic)도 포함되어 있습니다. 이 도식을 보면 AlphaFold가 MSA 정보와 쌍(Pair) 정보를 어떻게 통합하고 반복적으로 개선하는지 한눈에 알 수 있어요. 이전 모델들이 두 정보를 분리해서 처리했다면, AlphaFold는 이 두 정보를 'Evoformer'라는 핵심 모듈을 통해 끊임없이 상호 참조하고 정제하는 방식으로 구조 정보를 뽑아냅니다. 이 그림들은 단순한 삽화가 아니라, AlphaFold가 왜 그토록 정확한 예측을 할 수 있었는지에 대한 기술적인 해답을 담고 있다고 봐야 해요. 저자들은 이 시각 자료들을 통해 복잡한 딥러닝 구조를 이해하기 쉽게 제시하고 있답니다.
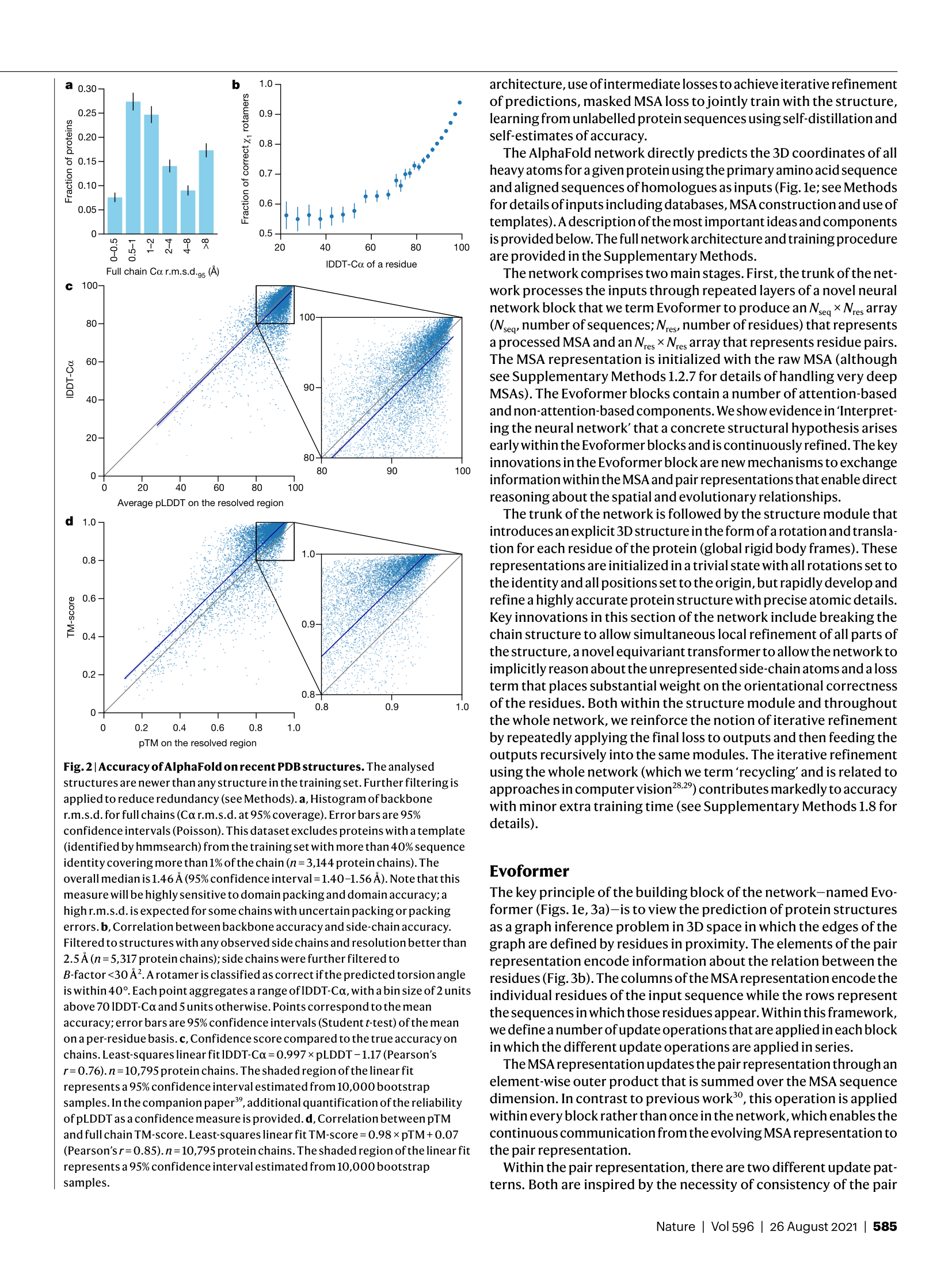

결과적으로, 논문 속의 그림과 도표들은 AlphaFold가 기존의 단백질 구조 예측 방법론들을 단순한 개선의 차원이 아니라, 질적으로 다른 수준으로 끌어올렸다는 것을 시각적으로, 그리고 수치적으로 명확하게 증명하고 있어요. 특히 CASP14 데이터셋 외에도, AlphaFold의 학습 데이터 마감일 이후에 PDB에 등록된 독립적인 검증 데이터셋(Recent PDB set)을 대상으로도 경쟁자들을 월등히 능가하는 성능을 보여주었다는 점은, 이 모델이 특정 데이터셋에 과적합되지 않은, 진정한 일반화 능력을 갖추고 있다는 것을 증명하는 결정적인 근거가 됩니다. 이 논문의 결과 섹션은 정말 과학적 성과를 데이터로 논증하는 교과서 같은 부분이라고 평가하고 싶어요.
5. 마법이 아닌 과학: AlphaFold 2의 핵심 방법론 심층 탐구 (Methods)
결과가 아무리 좋아도, 그 결과를 도출한 방법론이 핵심이겠죠. 논문의 Methods(방법론) 섹션은 바로 이 AlphaFold의 혁신적인 아키텍처와 학습 과정을 자세히 설명해 줍니다. 제가 이 부분을 읽으면서 가장 인상 깊었던 건, 생물학적 통찰력과 최신 딥러닝 기술을 정말 절묘하게 결합했다는 점이에요. AlphaFold의 아키텍처는 크게 두 가지 핵심 요소로 나눌 수 있습니다: Evoformer와 Structure Module입니다.
첫 번째, Evoformer(Evolutionary Transformer)는 단백질의 진화 정보를 담고 있는 MSA(다중 서열 정렬) 표현과 아미노산 쌍(Pair) 표현을 반복적으로 상호작용시키면서 개선하는 역할을 합니다. 이전 방법들은 MSA에서 얻은 쌍별 거리 제약을 초기 예측에 한 번 사용하고 끝냈지만, AlphaFold는 이 두 정보를 반복적인 어텐션(attention) 메커니즘을 통해 끊임없이 정제하고 있어요. 특히 이 Evoformer는 O(N²) 의 복잡도를 O(N·M)으로 효율화했는데, N은 잔기 수(Length), M은 MSA 항목 수(Depth)를 의미합니다. 이 복잡도 최적화 덕분에 수백에서 수천 개의 잔기를 가진 큰 단백질도 효과적으로 처리할 수 있게 된 것이죠. 저는 이 Evoformer가 AlphaFold의 '지능'의 핵심이라고 생각해요. 진화적 정보를 단순한 통계가 아닌 깊이 있는 생물학적 언어로 번역해내는 모듈인 셈이죠.
두 번째 핵심 요소는 Structure Module(구조 모듈)입니다. Evoformer를 거쳐 정제된 MSA와 쌍 표현을 입력으로 받아, 실제로 단백질의 3차원 구조를 생성하는 역할을 담당합니다. 이 모듈은 단백질 주쇄(backbone)의 각 아미노산 잔기에 대해 회전 행렬과 병진 벡터(Rotation and Translation)를 예측해요. 이 방식은 기존의 카테시안 좌표(Cartesian coordinate)를 직접 예측하는 방식보다 물리적 실현 가능성(physical realizability)이 높은 구조를 생성하는 데 유리하다고 저자들은 설명합니다. 즉, 단백질 구조가 가질 수 있는 물리적 제약을 모델 스스로가 학습하고 반영하도록 설계한 것이죠.
이 두 모듈을 연결하는 과정에는 'Recycling(재활용)' 메커니즘이 도입됩니다. Structure Module에서 생성된 3차원 구조에 대한 정보는 다시 Evoformer로 피드백되어 MSA와 쌍 표현을 한 번 더 개선하는 데 사용됩니다. 이 반복적인 정제 과정(Iterative refinement)이 AlphaFold의 정확도를 비약적으로 높인 핵심 비결이라고 저는 확신해요. 예측이 한 번에 끝나는 것이 아니라, 예측된 구조를 바탕으로 다시 진화적 정보를 개선하고, 이 개선된 정보로 다시 더 좋은 구조를 예측하는 과정을 여러 번 거치게 되니, 모델이 점점 더 단백질의 본질적인 구조적 특성을 깊이 이해하게 되는 거죠.
학습 과정에서도 혁신적인 요소가 발견되는데, AlphaFold는 PDB 데이터에 대한 지도 학습(Supervised learning)만으로도 높은 정확도를 달성했지만, 여기에 비지도 학습(Unsupervised learning)의 요소와 물리적 모델링의 특징을 융합하여 정확도를 더욱 향상시켰다고 논문은 설명합니다. 특히 손실 함수(Loss function)를 설계할 때, 단백질 구조의 모든 원자 간의 거리 오차(FAPE, Frame Aligned Point Error)를 최소화하는 방식으로 학습을 유도한 것이 원자 수준 정확도를 달성하는 데 결정적인 역할을 했을 거예요. 이처럼 AlphaFold의 방법론은 단백질 구조 예측의 물리적 경로와 진화적 경로를 딥러닝이라는 그릇 안에 완벽하게 융합시킨 결과물이라고 평가할 수 있습니다. 정말이지 대단한 설계 능력이라고밖에는 말할 수가 없네요!
6. 거인의 어깨 위에서: AlphaFold의 방대한 참고문헌과 학술적 배경
논문의 마지막 섹션 중 하나인 참고문헌(References) 목록을 보면, AlphaFold가 하루아침에 뚝딱 나온 기술이 아니라 수많은 선행 연구 위에 견고하게 구축된 결과임을 알 수 있어요. 논문에는 단백질 접힘 문제에 대한 초기 연구부터, 진화적 상관관계 분석(Co-evolution), 그리고 최근의 딥러닝 기반 구조 예측 방법론에 이르기까지 방대한 학술적 배경을 포괄하는 참고문헌들이 포함되어 있습니다. 저자들은 이 참고문헌들을 통해 AlphaFold가 기존 연구의 한계를 어떻게 돌파했는지, 그리고 어떤 아이디어를 가져와서 발전시켰는지 명확히 보여주고 있죠.
특히 진화적 상호작용 프로그램에 대한 참고문헌들이 눈에 띄는데, 이는 다중 서열 정렬(MSA)을 이용한 공진화(co-evolution) 분석이 AlphaFold의 핵심 입력 정보이기 때문입니다. Coupled-site analysis나 Direct Coupling Analysis (DCA)와 같은 기법을 통해 단백질 서열 내에서 상호작용하는 잔기 쌍을 찾아내려는 오랜 노력들이 있었고, AlphaFold는 이러한 진화적 제약 조건을 딥러닝 모델의 초기 입력으로 활용하여 예측의 정확도를 획기적으로 높인 것이죠. 저자들은 이처럼 선배 연구자들이 쌓아 올린 지식을 바탕으로 자신들의 혁신을 이끌어냈음을 참고문헌을 통해 겸손하게 인정하고 있답니다.
또한, 딥러닝(Deep Learning) 및 트랜스포머(Transformer) 아키텍처에 관련된 참고문헌들도 매우 중요한 비중을 차지하고 있어요. Evoformer라는 혁신적인 모듈 이름에서 알 수 있듯이, AlphaFold는 자연어 처리(NLP) 분야에서 큰 성공을 거둔 트랜스포머 구조의 장점을 생물정보학 데이터에 맞게 성공적으로 변형하여 적용했어요. 서열 정보(MSA)와 쌍 정보(Pair representation) 사이의 복잡한 관계를 모델링하는 데 어텐션 메커니즘을 사용한 것은, 정보 통합 및 특징 추출 능력을 극대화한 결과라고 볼 수 있죠. 이처럼 다른 분야의 최신 기술을 과감하게 차용하고 생물학적 문제에 맞게 최적화한 저자들의 통찰력에 다시 한번 감탄하게 됩니다.
이러한 방대한 참고문헌 목록은 AlphaFold의 성공이 단순히 엄청난 컴퓨팅 자원을 투입한 결과가 아니라, 수십 년간 축적된 생물학, 물리학, 컴퓨터 과학 분야의 지식을 총체적으로 융합한 결과라는 것을 명확하게 보여줍니다. 저자들은 논문의 끝부분에 위치한 참고문헌 섹션조차도 자신들의 연구가 서 있는 학술적 토대를 명시하는 중요한 역할을 하도록 배치하고 있어요. 저는 이 참고문헌들을 보면서, 진정한 혁신은 기존 지식에 대한 깊은 존중과 이해에서부터 시작된다는 사실을 다시 한번 깨달았습니다. 단백질 구조 예측 분야에 관심 있는 분들이라면, AlphaFold의 참고문헌 목록을 따라가 보는 것만으로도 이 분야의 역사와 발전 과정을 훑어볼 수 있을 거예요. 정말 가치 있는 참고 자료라고 생각합니다!

7. AlphaFold의 심장, Evoformer와 Structure Module의 협업 원리
앞서 방법론 섹션에서 AlphaFold의 핵심 구성 요소인 Evoformer와 Structure Module을 간략하게 설명드렸는데요. 이 두 모듈이 어떻게 유기적으로 협력하여 원자 수준의 정확도를 달성하는지 좀 더 깊이 파헤쳐 보는 것이 좋을 것 같아요. 솔직히 이 메커니즘을 이해하는 것이 AlphaFold의 진정한 혁신을 이해하는 열쇠라고 저는 생각합니다.
Evoformer는 단백질 구조 예측에서 가장 중요한 두 종류의 정보, 즉 MSA(다중 서열 정렬) 특징과 잔기 쌍(Pair) 특징을 동시에, 그리고 반복적으로 처리하는 모듈이에요. Evoformer 블록 내에서는 두 가지 중요한 어텐션 과정이 일어납니다. 첫째는 MSA 내의 각 서열 간의 정보 교환을 담당하는 어텐션입니다. 이를 통해 모델은 진화적 정보를 전체 서열의 맥락 속에서 이해하게 되죠. 둘째는 MSA 정보와 잔기 쌍 정보 간의 상호작용을 가능하게 하는 어텐션입니다. 이 두 정보가 끊임없이 서로에게 피드백을 주면서, Evoformer는 최적의 거리 행렬(Distance Matrix)을 유추해낼 수 있는 상태에 도달하게 되는 것이죠. 논문에서는 이 과정을 수많은 Evoformer 블록을 쌓아 올려 매우 깊은 신경망(Deep Network)을 구성했다고 설명합니다.
Evoformer를 통과한 정제된 쌍 특징은 이제 Structure Module로 전달되어 3차원 구조를 예측하는 데 사용됩니다. Structure Module의 가장 독특한 점은 단백질 구조를 좌표가 아닌 프레임(Frame)으로 표현한다는 사실이에요. 각 잔기에 대해 국소적인 3차원 프레임을 정의하고, 이 프레임이 다음 잔기의 프레임으로 어떻게 이동하고 회전해야 하는지를 예측하는 방식이죠. 이 방식은 단백질 주쇄를 강체(Rigid body)의 연속으로 모델링함으로써, 예측된 구조가 물리적으로 불가능한 모양이 되는 것을 방지하는 효과가 있어요. 이것이야말로 생물학적 지식을 모델 설계에 성공적으로 통합한 대표적인 예라고 볼 수 있답니다.
그리고 이 두 모듈을 엮어주는 'Recycling' 메커니즘을 빼놓을 수 없죠. Structure Module이 예측한 3차원 구조는 다시 새로운 쌍 특징(Geometric features)으로 변환되어 Evoformer의 입력으로 재활용됩니다. 마치 예측 결과를 보면서 '아, 내가 여기서 이런 정보를 놓쳤네. 다시 진화적 정보를 확인해 봐야겠어'라고 생각하는 것처럼, 모델 자체가 자기 수정적인 반복 과정을 거치게 되는 거예요. 이 Recycling 과정이 여러 번 반복될수록, 최종 예측 구조의 정확도는 급격히 상승하게 됩니다. 저자들은 이 반복 횟수가 정확도에 미치는 영향을 데이터로 보여주며, 이 메커니즘이 AlphaFold 성공의 결정적인 요소임을 분명히 하고 있답니다.
이러한 Evoformer, Structure Module, 그리고 Recycling의 삼위일체는 AlphaFold를 기존의 딥러닝 모델들과 구별 짓는 가장 큰 특징이자, 원자 정확도를 달성한 기술적 비결이라고 할 수 있어요. 복잡한 단백질의 구조 예측 문제를 정보 처리, 공간적 모델링, 그리고 반복적 정제라는 3단계로 완벽하게 분해하고 해결한 저자들의 접근 방식에 정말 깊은 존경심을 표할 수밖에 없네요. 이 아키텍처는 앞으로 생물학뿐만 아니라 다른 복잡한 데이터 모델링 분야에도 큰 영감을 줄 것이라고 확신합니다.
8. 새로운 구조 예측의 시대: AlphaFold의 임팩트와 후속 활용 방안
AlphaFold 논문의 결과가 발표된 후, 생물학계는 문자 그대로 '혁명'을 맞이했습니다. 논문에서도 언급하고 있듯이, AlphaFold는 이미 실험적인 단백질 연구에 그 유용성을 입증하고 있어요. 과거에는 구조를 알기 위해 몇 년을 기다려야 했지만, 이제는 AlphaFold를 통해 단 며칠, 심지어 몇 시간 안에 고도로 정확한 구조 예측을 얻을 수 있게 되었답니다. 이것은 실험의 계획과 방향 설정에 엄청난 가속도를 붙여줍니다.
저자들이 생각하는 가장 큰 임팩트 중 하나는 단백질 구조 데이터베이스의 확장입니다. 현재 PDB에 등록된 구조는 10만 개 정도지만, UniProt에 있는 수많은 단백질 서열 중 상당수를 AlphaFold로 예측할 수 있게 되었어요. 실제로 논문 발표 이후, AlphaFold 팀은 수많은 종의 거의 모든 단백질 구조를 예측하여 AlphaFold Protein Structure Database를 공개했고, 이는 전 세계 연구자들이 자유롭게 활용할 수 있게 되었습니다. 저자들은 이처럼 대규모 구조적 커버리지를 확보함으로써, 인류가 단백질 기능에 대한 이해를 넓히고 질병 치료, 신약 개발, 지속 가능한 에너지 솔루션 등에 이바지할 수 있다고 확신하고 있습니다.
AlphaFold 예측 구조는 실험 구조 결정의 초기 단계(Phase determination in X-ray crystallography X선 결정학에서의 위상 결정)에 분자 대체(Molecular Replacement) 모델로 사용될 수 있음이 논문에서도 언급되고 있어요. 이뿐만 아니라, 단백질 상호작용 연구나 돌연변이 효과 예측 등 다양한 생물학적 질문에 대한 가설 수립 및 실험 설계의 기반을 제공합니다.
물론 저자들은 AlphaFold의 한계와 미해결 과제에 대해서도 솔직하게 언급합니다. 예를 들어, 다중 단백질 복합체(Multi-protein complex)의 구조 예측이나, 리간드(ligand)나 보조 인자(cofactor)와의 결합 상태, 그리고 동적(dynamic)인 단백질의 움직임 등은 여전히 도전적인 영역으로 남아있어요. AlphaFold가 예측한 구조는 기본적으로 단백질의 가장 안정한 상태(Lowest energy conformation)에 가깝지만, 실제 단백질은 끊임없이 움직이고 환경에 따라 구조를 바꾸기 때문에, 이 동역학적인 측면을 포착하는 것은 다음 세대 모델의 숙제가 될 것 같습니다.
저는 이 논문을 읽으면서 AlphaFold가 단백질 구조 예측의 '검은 상자(Black box)'를 어느 정도 해소하는 데 기여했다는 점도 중요한 임팩트라고 생각했어요. Evoformer와 Structure Module의 설계 자체가 생물학적/물리적 원리를 반영하고 있기 때문에, 단순히 '좋은 결과가 나왔다'가 아니라 '왜 좋은 결과가 나왔는지'에 대한 설명 가능성(Explainability)을 높여주었거든요. 앞으로 이 아키텍처를 기반으로 한 후속 연구들이 단백질 접힘 원리에 대한 우리의 근본적인 이해를 더욱 심화시켜 줄 것이라 기대합니다. AlphaFold는 이미 생물학 연구의 패러다임을 '실험 중심'에서 '계산 및 예측 병행'으로 바꾸고 있는 중이라고 봐야 할 것 같아요. 정말 눈부신 미래가 우리 앞에 펼쳐져 있는 것 같습니다!
9. AlphaFold 2, 핵심 성과 한눈에 보기 (요약 카드)
지금까지 방대한 AlphaFold 논문 내용을 함께 살펴보셨는데요, 복잡한 내용을 마지막으로 핵심만 콕 집어 요약해 드릴게요. 이 카드 하나만 기억해 두셔도 충분할 겁니다!
자주 묻는 질문
논문을 읽고 나면 자연스레 생기는 질문들이 있죠. 저도 그랬어요! AlphaFold와 관련된 핵심적인 궁금증들을 Q&A 형식으로 정리해 보았습니다.
지금까지 존 점퍼의 'AlphaFold' 논문에 대한 저의 학습 노트를 함께해 주셔서 정말 감사합니다. 이 논문은 생명 과학의 미래를 바꿀 거라는 제 확신이 더욱 커지는 계기가 되었어요. 단백질 구조 예측 문제가 사실상 해결되었다는 저자들의 주장에 저도 고개를 끄덕일 수밖에 없었답니다.

'Tech & Science[기술과 과학] > AI & CompSci' 카테고리의 다른 글
| 과학의 제4 패러다임: 벤지오 논문으로 보는 AI와 과학의 결합 (2) | 2026.01.26 |
|---|---|
| AI 역사상 가장 중요한 논문 재조명: 홉필드 네트워크의 헤비안 학습과 안정 상태 (1) | 2025.12.19 |
| AI 시대 역량 키우기: '초예측: 세계 석학 8인에게 인류의 미래를 묻다' , AI가 바꿀 3가지 세상! (1) | 2025.10.02 |
| 99%가 모르는 AI의 역설: 닉 보스트롬 슈퍼인텔리전스에서 찾은 AI 시대 해법 (1) | 2025.09.20 |
| 컴퓨터 구조 책 추천:존 헤네시와 데이비드 패터슨, 그들의 고전 <컴퓨터 구조 및 설계> (0) | 2025.09.08 |



